you are here: home > newsletters > 04-2000 > DSEB newsletter
Division of Systematic and Evolutionary Biology (DSEB) - Spring 2000 Newsletter
- Message from the Chair
- Message from the Program Officer
- Message from the Secretary
- Phylogenetics for Dummies I: Optimization (Character Mapping)
Message from the Chair
Paula M. Mabee
Great to see many members of the Division of Systematic and Evolutionary Biology at the Atlanta meeting! DSEB co-sponsored two excellent, well-attended symposia and one workshop. Many thanks to Don Swiderski from DSEB, who with the Division of Vertebrate Morphology and the Society of Vertebrate Paleontology, organized the symposium "Beyond Reconstruction: Using Phylogenies to Test Hypotheses of Vertebrate Evolution." Speakers in this symposium demonstrated the many ways in which phylogenetic hypotheses can be used to examine and rigorously test ideas about patterns and processes in vertebrate evolution.
DSEB also co-sponsored the symposium, "Evolutionary Developmental Biology: Paradigms, Problems, and Prospects," co-organized by Richard Burian (DICI), Scott Gilbert (DDCB), Billie Swalla (DEDB) and me. This symposium was organized to inaugurate the formation of the new Division of Evolutionary Developmental Biology. The talks provided an intellectually exciting overview of this new interdisciplinary area.
DSEB sponsored a workshop entitled "Phylogenetics for Dummies." Anne Maglia and Don Swiderski gave a brief overview of phylogenetics and then, using handouts and overheads, went through the mechanics of character mapping, optimization and interpretation of patterns of character state change. The workshop was extremely well-attended by (70+) intelligent "dummies" from virtually all divisions of SICB plus at least one journalist. We intend to respond to this interest from SICB by organizing a workshop of this sort annually.
Our business meeting at Atlanta was well attended. Of the 267 SICB members who listed DSEB as one of their two member divisions, 35 chose DSEB for their name tag, and 18 people came to the business meeting. At the business meeting I expressed concern that few people were willing to run for divisional offices, and several members volunteered on the spot. Others expressed interest in organizing symposia for future meetings. This show of divisional interest and support is important. Our division is small, yet the contribution of systematics to the "integrative" activity of SICB is enormous, as demonstrated by the incorporation of phylogenies in many of the oral and poster presentations from all divisions. Our division is a perfect forum for systematists and other evolutionary biologists to generate and address questions concerning the interplay between developmental, paleontological, morphological, ecological, functional and molecular data sets.
We still need more members, and I encourage you all to sponsor the first year of membership for your graduate and postdoctoral students. If you have ideas for symposia, please don't hesitate to let us know. If you are interested in any of the offices for the division, contact us for more information. We welcome all interested in the integration of systematics with the rest of comparative biology.
Message from the Program Officer
Jon L. Norenburg
As of the Atlanta meetings, DSEB qualifies as the Stealth Division! We were everywhere, yet we were not easy to see. There was wide agreement that serious, if not always entirely correct, use of formal systematics was in evidence across the full spectrum of contributed paper and poster sessions. In addition, we sponsored and co-sponsored two very well-attended symposia—"Beyond Reconstruction: Using Phylogenies to Test Hypotheses About Vertebrate Evolution," superbly organized by Donald Swiderski, and "Evolutionary Developmental Biology: Paradigms, Problems and Prospects," co-organized by a bunch of people—including our Paula Mabee (who, in welcoming Evo/Devo to SICB, tactfully but clearly put them on notice that the phylogeny police will be watching ;-)
Presumably you all noticed a variety of changes in the abstract process and in organization of the meeting, which is part of why DSEB was less visible. Papers tended to be grouped more by function (ecology, comparative anatomy, behavior, etc.). We rarely have talks these days that focus on systematic theory. Although many of us enjoy a session of varied systematics talks, we also had a difficult argument against not having talks divided among sessions—where they are more likely to attract the attention of relevant non-systematists. This was the first year for the new program format, which inevitably introduced some unanticipated problems. Obviously, no solution makes everyone happy; achieving consensus on major changes is difficult and not without risk to our membership numbers. However, there was surprisingly wide support for the new program format at most of the business meetings from which I heard. You should let your officers and the society program officer know about specific problems encountered. The outstanding regret this year was the lack of competition for Best Student Paper, especially when compared with last year's outstanding showing. We were unable to justify a competition this year. There were some problems in other divisions this year, with people selecting one division to be identified with, but then selecting a different division to compete for Best Student Paper. That will not be permitted in the future.
SICB is now accepting proposals for symposia at Anaheim (2002), with approximately a mid-March to April 2000 deadline. You must consult with program officers before indicating divisional sponsorship. Informal symposia or workshops that are not intended for publication are also welcome. These can be very useful but are low on stress for the organizers. There is a strong feeling developing among the small core of people coming to the annual DSEB business meeting that frequent methods workshops and refreshers may be one of the most valuable things that we can do for the society. Don Swiderski and Anne Maglia hosted such a workshop this year, which ended up oversubscribed despite being left off the program and competing with evening events. SICB is also proposing significant support for very high-profile, innovative program activities.
Taxon Tooth shape 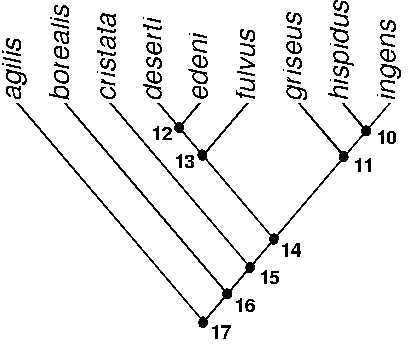
agilis 0 borealis 0 cristata 0 deserti 0 edeni 0 fulvus 0 griseus 1 hispidus 1 ingens 1 0 = rectangular
1 = square
First draw the tree. [Open MacClade, choose "new." Move the cursor to the bottom of the box that contains the question mark; hold down the mouse button and drag the box down to the number of taxa in the phylogeny. Fill in taxon names by selecting empty boxes and typing the names. To draw the tree, select "Go to Tree Window" in the "Display" pull-down menu. In the pop-up window warning that there no trees are stored, choose "Default Bush." Access the drawing tools by choosing the "Tools" menu (you may need to drag the toolbox onto the active screen). The only tool you need should be the "Move Branch" tool, in the upper right of the toolbox (the arrow). After selecting this tool, place the arrow icon on a branch (ingens) and drag it to any other branch (hispidus) to make a dichotomy. (You may need the other tools to draw more complex trees; when you select a tool, a description of its function is in the text box at the bottom of the toolbox. The "Collapse Branch" or "Collapse Clade" tools are especially useful for drawing polytomies.)]
Enter the character data. [Return to the data entry window by choosing "Go to Data Editor" on the "Display" pull-down menu. Click on the data boxes (which, by default, are filled with question marks) and enter the character state code, 0 or 1. We use 0 and 1 because the program needs the data in numerical form to compute the number of changes. You may also want to add character names in the boxes below the character numbers. Choose "State Names" in the "Display" pull-down menu, which will give you a pop-up window in which you can add long labels for the traits and detailed descriptions of the states.)]
Trace the character on the tree. [Go back to the tree window and choose "Trace Character" on the "Trace" pull-down menu.] The tree will change colors (or patterns), and a dialog box will appear in the lower right-hand corner of the window that tells you there is only one change, from 0 to 1 (from rectangular to square).
Optimization begins at the top of the tree and assigns each internal node the state shared by the two nodes immediately above it. (The internal nodes, representing hypothetical ancestors, are not labeled by MacClade.) In the first pass, Nodes 10 and 12 are assigned State 1; Nodes 11 and 13 are assigned State 0. Node 14 could be assigned either 1 or 0 because one descendant of Node 14 has State 0 and the other descendant has State 1. For now, this is left unresolved, and the next node (15) is considered. Because State 0, the rectangular tooth, is found in both lineages derived from Node 15, it is more parsimonious to infer that the ancestor represented by this node has the rectangular tooth. If this ancestor had the square tooth, then the rectangular tooth would have arisen twice. This is not the most parsimonious interpretation possible for two states, which imply only one transformation. Were the square tooth assigned to Node 15, that would imply a second transformation, for which we do not have evidence. In contrast, assigning the square tooth to Node 15 implies only one change, from square to rectangular (other nodes are assigned State 0). The second pass resolves the ambiguity by taking into account the node below, as well as the nodes above. So, the 0 below and the 1 and 0 above are most parsimoniously explained by assigning State 0 to Node 14.
Not every character can be unambiguously resolved at every node. To create a data set in which this problem arises, edit the data matrix so that cristata has State 1. [Return to the data editor window, click on the cell and type the new number, return to the tree window]. By default, MacClade will indicate with hash marks that there is more than one way to optimize the trait on some branches (and the nodes just above those branches). You need to tell MacClade how to treat ambiguous nodes. [Choose "Resolving Options..." on the "Trace" pull-down menu. Select the option that will show all states, if it is not already selected]. Trace the character again, and you will see that there are now two ambiguous nodes (14 and 15) because there are two ways to optimize them, and both imply the same number of changes. To see one of the choices, return to the "Resolving Options…" window and select Acctran (accelerated transformation), and then trace the character, again. The change is attributed to the earliest possible ancestor, so Nodes 14 and 15 are assigned the square tooth, and the transformations are 0 to 1 to 0. To see the other choice, select Deltran (delayed transformation). The change from rectangular to square is attributed to the latest possible ancestor, so Nodes 14 and 15 are assigned the rectangular tooth, and there are two changes from 0 to 1. Notice that Acctran favors reversals to the primitive state (or losses of the derived state) and Deltran favors multiple gains of the derived state. No matter how the character is optimized in the second example, we are forced to conclude that there are two changes in tooth shape even though we recognized only two shapes.
For the third example, edit the data matrix so that agilis, borealis and cristata have State 0; deserti, edeni and fulvus have State 1; and griseus, hispidus and ingens have State 2. 0 = triangular, 1 = square, 2 = rectangular. This is an example of a multistate character; the previous examples were two-state or binary characters. Multistate characters can be analyzed as ordered or unordered. [Go to the "Display" menu and select "character type." This will display a list of characters, types and number of changes. The default is unordered. Click on the character, then go to the "Assume" pull-down menu and select "Character type"]. Selecting ordered implies an assumption that changes between 0 and 2 must have gone through 1. The unordered option leaves open the possibility of a direct change between 0 and 2 without going through 1. Select unordered and Deltran. Node 14 is assigned State 0 because the changes to 1 and 2 are both delayed and it is possible to go from 0 to 2 without going through 1. Now select ordered. Node 14 will be assigned State 1 (choice of Acctran, Deltran or neither does not matter in this case).
In more complex cases, different combinations of ordered or unordered with Acctran or Deltran can lead to quite different interpretations of the number of changes and the lineages in which those changes occurred. To see how, edit the data matrix so that agilis, deserti, edeni and fulvus have state 0; borealis and cristata have 2; and griseus, hispidus and ingens have 1; and then compare the results under the various combinations.
(C) What can be learned from optimization? As demonstrated by the examples given in the previous section, optimization can be used to trace histories of observed traits. When multiple traits are optimized, the results can be used to examine relationships among those traits. For example, optimizations of body weight and femur length can be used to investigate evolutionary allometry of femur length. When both traits are coded and analyzed unordered, the comparison of the two optimizations will indicate how often the two traits changed simultaneously and in the same direction. A similar approach could be used to investigate coordination of changes in functionally or developmentally related structures (e.g., tooth and jaw shapes, or neural crest derivatives). Optimization also could be used to test a hypotheses that an intermediate morphology was historically intermediate, or to test a hypothesis of adaptation, by asking whether a morphological change arose before or after its presumed cause (e.g., a shift to a new diet).
Features that would not normally be regarded as intrinsic traits can also be optimized, such as biogeographical distribution or geological age. To test a biogeographical hypotheses, the regions in which a taxon is found can be treated like a character. Should the tree suggest an implausible biogeographical hypothesis, it is possible to rearrange the tree so it is consistent with an alternative hypothesis and estimate the number of additional steps required. The hypotheses can then be compared, but it is important to take into account the relative quality of the data (e.g., reliability of the characters, completeness of taxon sampling, completeness of the fossil record).
Geological ages of taxa (e.g., time of first occurrence) can be optimized in several ways. The estimated ages of fossils can be input as quantitative values, or coded and optimized by any of the methods discussed above. In MacClade, it is also possible to type in names of geological periods and select stratigraphic in the character type list, which treats the information as an ordered character and also prevents reversal (so older taxa cannot be derived from younger taxa). Like the optimization of geographical data, optimization of geological age data may say something about the completeness of the fossil record or about the reliability of the characters used to infer the phylogeny.
These are only a few examples of the uses of optimization. Any evolutionary hypothesis can be recast in phylogenetic terms, and the expected optimization compared to that implied by the tree. The crucial point is that rejection or acceptance of the evolutionary hypothesis is contingent on acceptance of the phylogeny. A different phylogeny may lead to a different conclusion. The next workshop will consider the question of how to choose among different phylogenies presented in the literature.